
博客
如何安装 ColabFold 并在本地运行 AI 蛋白质折叠

- 蛋白质折叠问题
蛋白质在细胞中起着结构、机械、化学和信号传导的作用。设计或发现的蛋白质越来越重要;有效药物的生产是一个利润丰厚的市场,也带来了改变世界的潜力。
蛋白质,简单来说就是一串氨基酸。解决蛋白质是基于单独的氨基酸序列来确定 3D 结构。强制组合搜索和找到正确结构的困难在统计学上是不可能的,蛋白质结构预测长期以来一直被认为是徒劳的。至少是这样,直到60%的准确率 Alphafold1 击败了其他 CASP13 的竞争对手,随后在 CASP14 出现了接近90%的准确率的 AlphaFold2。
自2020年以来,AlphaFold2 的学术和开源替代品在结构生物信息学方面继续取得进展。我们想探索 AlphaFold 自 CASP14 以来在相关模型中的进展,并专注于 ColabFold,以及 ColabFold 如何通过改进绝对不那么光彩照人的多序列比对步骤来显著缩短蛋白质结构预测时间。最新的 AlphaFold2 将被寻址为 AlphaFold,而 CASP13 的第一次迭代将被寻址至 AlphaFold1。
- 什么是 ColabFold?
ColabFold 是一种蛋白质预测算法,主要用于谷歌的合作笔记本电脑环境。但是,尽管谷歌 Colab 可以进行小规模的探索性工作,但如果不能完全不变地访问 GPU 和 TPU 等高性能加速器,它在生产或认真研究方面存在局限性。
我们将逐步在您自己的预制机器上本地部署 ColabFold,并演示如何预测负责日本萤火虫生物发光的荧光素酶(蛋白质数据库中的 2d1s)的结构,而不是依赖谷歌 Colab。
- 为什么使用 ColabFold 而不是 AlphaFold
准确的蛋白质预测算法的发展,深深植根于利用深度学习训练来分析人眼所知的潜在行为的爆炸式发展。然而,我们想重点关注 AlphaFold、RoseTTAFold 和 OpenFold 都将其作为蛋白质结构预测配方的一部分使用的 MSA 模块。
多序列比对(MSA)是相似蛋白质(或 DNA)序列的集合,其排列方式是比较和评估来自不同样本的相似氨基酸序列。通过捕获共享相关结构的熟悉序列,模型可以访问并引用模板,以避免连续预测已知序列。尽管 MSA 非常有用,但构建大型 MSA 模板库在计算上可能很昂贵,并且可能无法很好地扩展到大型序列集。
ColabFold 结合了一种称为 MMSeqs2 的快速并行 MSA 算法,在预测序列所需的总时间上大大提高了标准 AlphaFold 的速度。虽然 ColabFold 并不独立,但它使用 AlphaFold 或 RoseTTAFold 作为结构预测模型,并将其与 MMSeqs2 相耦合。
ColabFold MSA 算法 - MSEqs2
ColabFold 论文的合著者 Martin Steinegger 助理教授领导了 MMSeqs2 的开发,他表示,使用 MMSeqs2 可以将构建 MSAs 的时间从几个小时缩短到几分钟,该论文声称可以加快40到60倍。
MSA 加速的关键是使用 k-mer 搜索进行预过滤的新策略(k-mer 是序列长度为 “k” 的氨基酸块)。MMSeqs2 在序列之间的一行中寻找具有多个共享 k-mer 的序列。由于 MMSeqs2 允许相似序列词之间的部分 k-mer 匹配,因此与原始 DeepMind AlphaFold 工作流和笔记本中使用的其他 MSA 搜索算法(如 jackhmer )相比,它可以使用更长的 k-mer,并且只损失一点灵敏度。
此外,由于 MMSeqs2 使用的序列数据库和比对本身具有实质性的存储器需求(例如,对于 MSA 中具有约3000万个序列的长度342的序列,约 80GB 的 RAM),因此 MMSeqs2 可以高速缓存用于重复查询的 MSA,以加快热门查询的响应时间。当运行笔记本预测我们的 2d1s 生物发光蛋白时,MMSeqs2 仅用了7秒就返回了 MSA,而不是60秒。想要将其序列保密的蛋白质工程师可以建立自己的服务器来运行 GPL 3.0 许可的 MMSeqs2。
- 为什么要在自己的机器上运行 ColabFold
虽然 ColabFold 是专门为在谷歌的 Colaborary 云笔记本环境中运行而开发的,但它的真正意图是让所有人都能使用蛋白质折叠。通过提供一个可访问云的蛋白质预测选项,使强大的软件民主化,使理论家能够进行实验。在本节中,我们将介绍如何使用 AlphaFold 来预测蛋白质结构,使用 ColabFold 和 MMSeqs2 进行快速结构预测。
云笔记本非常适合共享或展示工作。然而,对 GPU 和其他硬件的访问可能会受到可用性和使用上限的限制。
对于更深入的工作,再现性是关键,控制您的环境和所有依赖性是值得的,这也是联泰集群提倡研究机构选择预计算解决方案的原因。探索我们的各种高性能多 GPU 计算平台,从工作站到服务器再到集群。配置您理想的系统,咨询针对您工作负载有价值的工程师建议,并立即获得报价。
为了实现让更多人更容易获得高质量蛋白质折叠的目标,我们将通过本地机器运行 ColabFold。除了云笔记本提供的可访问性之外,为 MSAs引入MMSeqs2 的加速功能使 ColabFold 值得单独使用。从 MMSeqs2 获取 MSAs 仍然需要服务器调用,但 AlphaFold 在本地运行,也可以使用带有 RoseTTAFold 的 ColabFold 改进。
- 如何设置 ColabFold 在本地机器上运行
首先,我们将初始化并激活一个虚拟环境来管理 Python 中的依赖关系。我们使用 virtualenv,所以如果您使用 Anaconda 或 miniconda,您可能需要修改一些内容。然而,在 Python-m 中,venv 应该以几乎相同的方式工作。同样,我们将假设一个 Unix 环境,最好是您最喜欢的 Linux 发行版。如果您想使用 Windows(例如使用 Anaconda Unix shell),您可能需要进行一些实质性的调整才能使其正常工作。
要从命令行创建和激活虚拟环境,请执行以下操作:
virtualenv cl_env --python=python3.8
source cl_env/bin/activate
pip install jupyter notebook
这是 Rive Sunder 的这款分叉提供的 ColabFold 存储库的修改版本,笔记本电脑去掉了许多 Colab 特定的用户界面、安装命令和其他我们不会使用的部分。否则,原来的 ColabFold 存储库由 sokrypton(Sergey Ovchinnikov)链接到这里。
# To Use Modified/Stripped ColabFold
git clone git@github.com:rivesunder/ColabFold.git
# To Clone Original and Modify ColabFold on your Own
git clone git@github.com:sokrypton/ColabFold.git
cd ColabFold
pip install -q --no-warn-conflicts 'colabfold[alphafold-minus-jax] @ git+https://github.com/sokrypton/ColabFold'
pip install --upgrade dm-haiku
最有可能的是,上面的命令安装了没有 GPU 支持的 Jax 的基本版本。jax 0.4.13 和 jaxlib 0.4.13 是仅限 CPU 的版本——要启用 GPU 支持,您需要一个更新的版本,越新越好。您可以查看:
#pip freeze | grep jax
假设您想使用本地 GPU,您可以使用以下模式安装正确版本的 jax。CUDA11 安装在这台机器上,但您可以检查 JAX 文档以获得适合您的设置的正确版本。
# First, uninstall JAX
pip uninstall jax jaxlib
# Next, install JAX with CUDA
pip install --upgrade "jax[cuda11_local]" -f \\ https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
# Verify JAX Version is newer than 0.4.13
pip freeze | grep jax
接下来的几个命令改编自官方的 ColabFold 实现,以便在虚拟环境中创建与 AlphaFold 的符号链接。
ln -s cl_env/lib/python3.8/site-packages/alphafold alphafold
# This next command is not needed for the modified notebook. Otherwise:
#ln -s cl_env/lib/python3.8/site-packages/colabfold colabfold
还有一个 find-and-replace 命令,用于在 AlphaFold 文件中添加剪切函数。请确保以下命令在一行中。
sed -i 's/weights = jax.nn.softmax(logits)/logits=jnp.clip(logits,-1e8,1e8);weights=jax.nn.softmax(logits)/g' alphafold/model/modules.py
最后,我们可以编写一个文件,作为指示 ColabFold 已设置并准备就绪的标志:
touch COLABFOLD_READY
ColabFold 预测萤光素酶蛋白结构
如果一切都成功了,现在可以在命令行上键入 “jupyter 笔记本” 来启动本地 jupyter 服务器。导航到 ColabFold 文件夹中的 AlphaFold2.ipynb,导航到 Runtime 并点击 Run All(或 Ctrl+F9)运行所有细胞,下载 AlphaFold 参数并运行日本萤火虫萤光素酶 2d1s 的结构预测。
如果你想输入自己的序列,或者突变默认的萤火虫萤光素酶,你可以修改或替换笔记本第三个代码单元中分配给 “query_sequence” 的序列。
笔记本提供:
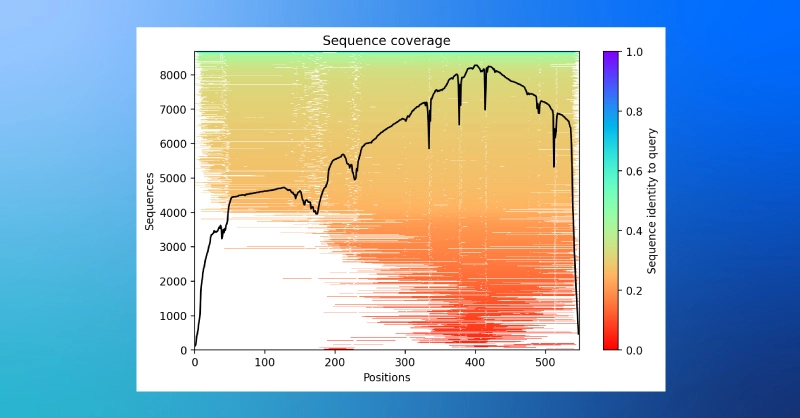
- 结构预测运行时 MSA 中的序列覆盖图。
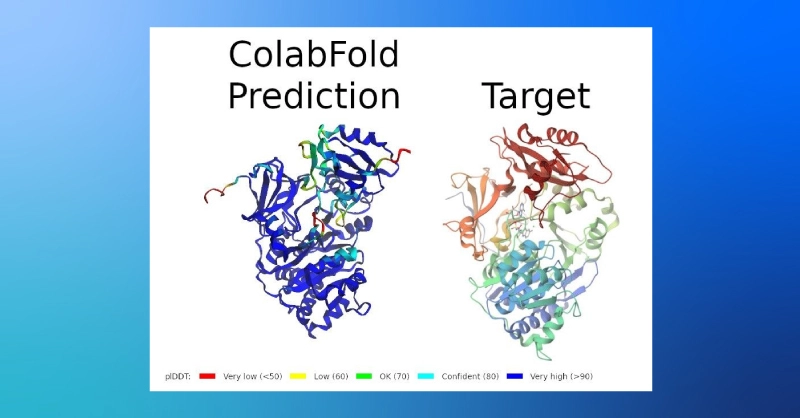
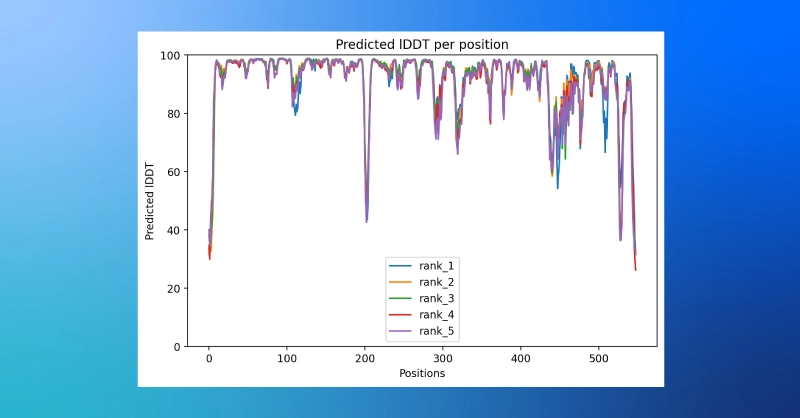
- 结果包括置信度预测、预测局部距离差测试(plDDT)、在下面的预测结构中进行颜色编码(目标颜色仅指示序列位置)。
- ColabFold 笔记本还可以提供按氨基酸位置划分的 plDDT 值图。
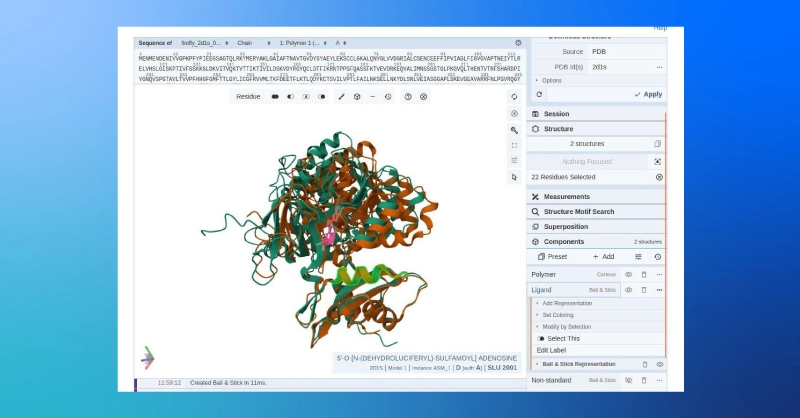
- 并排比较为我们提供了预测结构和目标结构的结构相似性的一般视图,但我们也可以使用 PDB 的在线结构查看器来比较对齐的结构。
- 人工智能和蛋白质折叠的下一步是什么?
在2020年 CASP14 之后阅读新闻报道时,蛋白质折叠爱好者认为蛋白质折叠问题已经没有什么可解决的了,这是情有可原的。事实上,蛋白质结构预测只是蛋白质折叠问题的一部分。翻译后修改、折叠伴侣和蛋白质糖基化是可以改变给定蛋白质序列的结构和化学功能的几种机制。
自 CASP14 以来的几年里,进展一直在继续。已经开发了具有可比性能的其他蛋白质结构预测模型。RoseTTAFold 和 OpenFold 使用 MSA,而 ESMFold 和 OmegaFold 在不首先构建 MSA 的情况下从单个序列预测结构。
一个热情的社区出现了,他们应用黑客思维,使用 AlphaFold 和同行模型对多聚体和蛋白质结合物等进行建模。在 CASP15,多聚体蛋白质和蛋白质-蛋白质相互作用的建模结构有了实质性改进,这是 CASP14 AlphaFold 的弱点。目前发布的 AlphaFold 模型于2023年年中发布,其特点是提高了对接、核酸、配体结合等方面的准确性,距离更准确的蛋白质预测又近了一步。虽然90%的置信度是相当准确的,但对于设计一种只有氨基酸序列的有目的的新蛋白质来说,它还不够准确。
AlphaFold 和随后的结构生物学和蛋白质工程模型的成果现在包括设计。ColabFold 的开发人员还建立了 ColabDesign,它可以使用蛋白质结构预测模型来提出合理的序列来结合靶标或拟合所需的结构。扩散去噪模型在图像/视频生成方面引起了人们的极大兴趣,扩散最近在蛋白质结构领域也取得了进展。贝克实验室及其同事开发的 RFDiffusion,以及蛋白质结构扩散模型的其他发展,可能预示着为治疗、材料科学和化学工程定制设计蛋白质的令人兴奋的可能性。
运行人工智能蛋白质预测对任何一台计算机来说都不是一项容易的任务,但在不依赖外部硬件来源的情况下保持数据接近,对于数据敏感的操作来说是有价值的。联泰集群为分子发现提供了高性能工作站、服务器和集群,所有这些都经过了最高标准的测试、优化和验证。今天就和我们的工程师谈谈,马上就可以配置你的 dream machine。




相关贴子
-
 技术分享
技术分享AlphaFold 更新显著提高了对接、核酸和 PTMS 的准确性
2024.04.12 14分钟阅读 -
 技术分享
技术分享如何加速工业产品设计中的原型制作
2024.12.13 33分钟阅读 -
 技术分享
技术分享机器学习和深度学习中需避免的 7 个常见错误与局限性
2025.11.08 34分钟阅读